The failure of the
common disease common variant paradigm has led to an effort to identify rare variants that may contribute to disease in human populations. To identify such rare variants, one needs to look at more individuals at denser genomic coverage. This has been one of the inspirations of the
1000 Genomes project, which aims to capture full genome sequences of more than the 1,000 individuals of its title.
A new paper has identified an important property of rare variants: they tend to be population-specific: rare variants in Europeans are mostly not the same as rare variants in Asians or Africans. A corollary of this observation is that a sample of X global individuals
is effectively much smaller than X for the purposes of identifying rare variants: you expect 1 in 1,000 individuals to possess an allele with 0.1% frequency, but if the allele occurs at 0.1% frequency in 250 Europeans, and at 0% frequency in 750 non-Europeans, then you expect to find 0.25 individuals in such a sample, in other words, you are very likely to miss it entirely.
The practical consequence of this, is to identify most rare variants in humans you need even larger samples: the 1000 genomes project is likely to miss many of them, and, correspondingly, a large part of the potential contribution of rare variants to disease. A second consequence is that
if the so-called
missing heritability is hiding in rare variants, different populations will get the same diseases but for different genomic reasons.
Was this unexpected?
This finding was not entirely unexpected. Consider the human species prior to its separation: it would harbor alleles ranging from very low to very high frequencies.
The very low-frequency alleles would have a high probability of being lost by drift in both the entire species (hence becoming irrelevant), or by founder effect in most human sub-populations. Only those that had some selective advantage might be expected to rise in frequency. Consequently, low-frequency alleles
prior to the separation of Homo sapiens into regional sub-populations would
mostly
not persevere as low-frequency alleles today.
How about very low frequency alleles that arose
after the separation of modern humans into Africans, Asians, Europeans, etc.? Again, the odds are stacked against those moving around: with a low migration rate between different human groups, it is fairly unlikely that a
rare allele would migrate. Consequently, rare alleles that arose in regional populations were either lost by drift, or they grew in frequency (becoming non-rare), and, if, they persevered as rare variants, it is unlikely that
they would be exchanged with other regional populations.
So, where do the rare variants mostly come from? They have to be mostly
recent variants that have not had the time to either go extinct due, or to become common by drift, and their rarity and young age makes it unlikely that they had the chance to emigrate to other regional populations.
Demographic estimates
The paper's other major contribution is in providing some fairly tight estimates of human demographic parameters, much better than previous ones. A summary can be seen on the left. What I especially like about this is that the authors explicitly tackle the very young age for the European-East Asian split,
without invoking a
"long hiatus" as previous papers did.
Here is their explanation:
The narrow confidence intervals on some of the parameters should not obscure the fact that the parameter estimates are model-dependent. As a simple example, a model that does not allow for migration would require more recent split times to produce similar levels of population divergence. The demographic history of the four populations considered is much more eventful than what is accounted for by our model. Additional geographically intermediate populations from the Near East and Central Asia that were not included in our analysis might contribute significantly to the allele frequency distribution as ghost populations (19).Incorporating an appropriate number of source populations for estimates of migration has been a general limitation of two-and three-population models under isolation migration coalescent, approximate Bayesian computation, and diffusion-based approaches. This limitation might explain why our estimate of the divergence between East Asians and Europeans is more recent than estimates based on archaeological evidence (18), but is comparable with estimates of 23 kya (20) under an approximate Bayesian computation approach and 25 kya under an isolation migration approach with mtDNA X and Y sequence data (21).
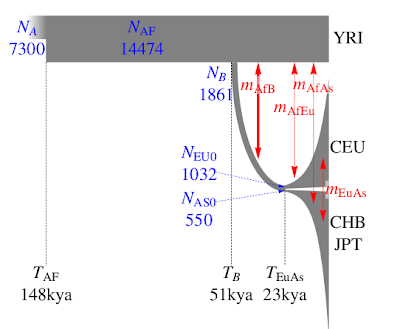
This makes excellent sense: we don't need to hypothesize that Proto-Eurasians spent tens of thousands of years in the Near East in a long stasis for no apparent reason; the archaeological record shows the arrival of anatomically modern humans in Europe long before 23,000 years ago. Indeed the earliest Europeans, and certainly those of 23,000 years ago already had typical Caucasoid physical characteristics. Hence, the idea that Europeans' ancestors were still marooned as undifferentiated Eurasians together with the ancestors of the Chinese difficult to swallow.
Instead, we can accept that East-West Eurasian differentiation began already shortly after Out-of-Africa, and the younger
apparent divergence of Europeans and East Asians is due to gene flow, perhaps from an unsampled population that contributed genes to
both.
Another interesting observation in the paper is that the divergence between Yoruba and Eurasians is a likely
under-estimate of the Out-of-Africa event. This makes excellent sense, as West Africans are not expected to be identical with Proto-Eurasians or their closest African relatives. Hence, the 51ky is an
over-estimate, and the true age seems to correspond well with the appearance of fully modern (in both appearance and behavior) Upper Paleolithic people in Eurasia, in full agreement with the
recent scenario I posted on the blog, the end of Marine Isotope Stage 3, and the coalescence of most modern human Y-chromosomes.
Indeed, even the age of the African population (148kya) seems to be consistent with the recent re-dating of
Y-chromosome Adam to 142kya. It would be interesting to include some African hunter-gatherers in the future; unfortunately, one of the great defects of the 1000 Genomes Project is its lack of African hunter-gatherers, which are, perhaps the
most interesting populations for human origins research.
In any case, it is refreshing to see a genetics paper that tries to show how the genetic dates may deviate from the archaeological ones for good
internal reasons, and seeks to explain the small discrepancies between the two by the limitations of genetic methods. This is much better than proposing archaeological implausible scenaria to "save" the genetic evidence, which, until this paper came along, did not really have the fairly tight confidence intervals necessary for genetic-archaeological correlations.
PNAS doi: 10.1073/pnas.1019276108
Demographic history and rare allele sharing among human populations Simon Gravel et al.
High-throughput sequencing technology enables population-level surveys of human genomic variation. Here, we examine the joint allele frequency distributions across continental human populations and present an approach for combining complementary aspects of whole-genome, low-coverage data and targeted high-coverage data. We apply this approach to data generated by the pilot phase of the Thousand Genomes Project, including whole-genome 2–4× coverage data for 179 samples from HapMap European, Asian, and African panels as well as high-coverage target sequencing of the exons of 800 genes from 697 individuals in seven populations. We use the site frequency spectra obtained from these data to infer demographic parameters for an Out-of-Africa model for populations of African, European, and Asian descent and to predict, by a jackknife-based approach, the amount of genetic diversity that will be discovered as sample sizes are increased. We predict that the number of discovered nonsynonymous coding variants will reach 100,000 in each population after ∼1,000 sequenced chromosomes per population, whereas ∼2,500 chromosomes will be needed for the same number of synonymous variants. Beyond this point, the number of segregating sites in the European and Asian panel populations is expected to overcome that of the African panel because of faster recent population growth. Overall, we find that the majority of human genomic variable sites are rare and exhibit little sharing among diverged populations. Our results emphasize that replication of disease association for specific rare genetic variants across diverged populations must overcome both reduced statistical power because of rarity and higher population divergence.
Link





